- Property & Casualty
- Life & Health
- Knowledge Center
-
About Us
About Us OverviewCorporate Information
TOP

Der Einfluss des Europarechts auf nationales Haftungsrecht wird in vielen Bereichen oftmals übersehen oder zumindest unterschätzt. Hierfür ist gerade die Medizinprodukthaftung ein gutes Beispiel. Europäische Normsetzung durch Richtlinien und Verordnungen sowie Entscheidungen des Europäischen Gerichtshofs zur Umsetzung dieser legislativen Maßnahmen in nationales Recht haben in den vergangenen Jahren das Haftungsregime im Medizinproduktebereich geprägt und verändert.
Dabei hat kaum ein Bereich in der medialen Auseinandersetzung eine größere Rolle gespielt als dieser; dafür haben in den vergangenen zehn Jahren viele Skandale gesorgt. Angefangen bei mit Industriesilikon gefüllten Brustimplantaten1 über sich abrasiv verschleißende Hüftköpfe bis hin zu Beckenbodennetzen, die sich regelmäßig lösten, sorgten Medizinprodukte immer wieder für negative Schlagzeilen. Nachdem bereits im Jahr 2012 Funktionsausfälle bei Herzschrittmachern aufgetreten waren,2 sorgte auch in diesem Jahr der Rückruf von mehr als 150.000 Herzschrittmachern für Aufsehen.3
Immer wieder stand deswegen das durch die Medizinprodukterichtlinie (Medical Device Directive – MDD)4 geregelte Zulassungsverfahren für den Einsatz von Medizinprodukten in der Kritik. Aus diesen Gründen hat die EU das Zulassungsverfahren überarbeitet und mit der Medizinprodukteverordnung (Medical Device Regulation – MDR)5 bereits im Jahr 2017 neu geregelt. Allerdings tritt diese Verordnung erst am 26. Mai 2020 in Kraft. Zunächst nur wenig beachtet, rückte diese Verordnung im November 2018 durch Veröffentlichungen des „International Consortium of Investigative Journalists – ICIJ“ wieder in den Mittelpunkt des Interesses, nachdem diese umfangreich über fehlerhafte Medizinprodukte berichtet hatten.6 Diese Berichterstattung führte sogar zu einer Anfrage im Deutschen Bundestag.7
All dies ist Grund genug für eine Bestandsaufnahme: Wie sieht das aktuelle Zulassungsverfahren für Medizinprodukte (nach der MDD) aus? Welchen Einfluss haben Maßnahmen der europäischen Legislative und Judikative auf das deutsche Haftungsrecht für Medizinprodukte? Welche Veränderungen und Konsequenzen bringt die neue Medizinprodukteverordnung (MDR) mit sich? Und ist die Verordnung geeignet, auch künftigen Entwicklungen in der Medizintechnik Rechnung zu tragen?
Obwohl der Schwerpunkt dieses Artikels auf der Darstellung der europäischen Rechtslage liegt, sind defekte Medizinprodukte bei weitem kein rein europäisches Thema. In den USA sind 32 Millionen Medizinprodukte im Einsatz, d. h. jeder zehnte US-Amerikaner ist derzeit mit einem Medizinprodukt ausgestattet. Der Zulassungsprozess der US-amerikanischen Food and Drug Administration (FDA) hat viele Ähnlichkeiten mit dem europäischen Verfahren und steht ebenfalls in der Kritik.
Alle Medizinprodukte müssen bei ihrem Inverkehrbringen gem. Art. 17 Abs. 1 MDD8 mit einer CE-Kennzeichnung versehen sein. Dies soll gewährleisten, dass sie kein Gesundheitsrisiko für den Patienten in sich bergen.9 Um ein solches CE-Zeichen insbesondere für Implantate10 zu erhalten, ist gem. Art. 11 MDD11 nach einer Konformitätsbewertung eine sog. EG-Konformitätserklärung entsprechend Anhang II der MDD erforderlich. Dieser Prozess wird gem. Art. 16 MDD durch die sog. Benannten Stellen durchgeführt.12 Hierzu ist für den Nachweis, dass das Medizinprodukt kein Gesundheitsrisiko darstellt, vom Hersteller eine „Klinische Bewertung“ vorzulegen, deren Inhalt durch Anhang X der MDD bestimmt wird. Entweder führt der Hersteller hierfür eine klinische Studie durch, oder er weist nach, dass es bereits vergleichbare zertifizierte Produkte gibt. Dieses sog. Äquivalenzprinzip ist der wesentliche Ansatzpunkt für die Kritik an der gegenwärtig geltenden Regelung, denn für den Nachweis der Vergleichbarkeit reicht ein wissenschaftliches Gutachten aus. Vereinfacht gesprochen führt dies dazu, dass eine Konformitätsbewertung faktisch nur nach Aktenlage erfolgen kann.
Den Ablauf dieses Verfahrens verdeutlicht das Schaubild 1.
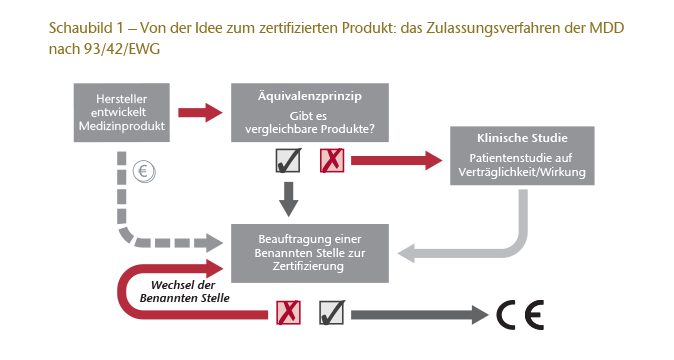
Weitere Kritikpunkte am gegenwärtigen Verfahren sind zum einen die Tatsache, dass die Benannten Stellen für die Zertifizierung vom Hersteller bezahlt werden und damit ein wirtschaftliches Interesse besteht, diese im Sinne des Auftraggebers durchzuführen. Zum anderen wird bemängelt, dass der Hersteller selbst dann, wenn eine Benannte Stelle keine Zertifizierung erteilt hat, problemlos so oft zu einer anderen Stelle wechseln kann, bis letztlich die erforderliche Konformitätserklärung erteilt wird.
Dennoch machen die Verantwortlichkeiten nach der MDD die Benannten Stellen nicht zu Überwachungsstellen. Vielmehr ist ihre Beziehung zum Hersteller ausschließlich privatrechtlich; sie haben lediglich die Rolle des Partners/Begleiters im Zertifizierungsverfahren. Die Verhinderung einer vorsätzlichen Umgehung der gesetzlichen Vorgaben ist die Aufgabe der nationalen Überwachungsbehörden.13
Entgegen der mit dem CE-Zeichen unbestritten verbundenen Konnotation eines „auf Herz und Nieren“ geprüften Produkts führt dieses Verfahren im so sensiblen Medizinproduktebereich mithin zu dem Ergebnis, dass viele mit diesem Zeichen versehenen Produkte nicht wirklich konkret geprüft worden sind.
Gerade der oben erwähnten Fall der mit Industriesilikon gefüllten Brustimplantate mit weltweit mehr als 400.000 potenziellen Anspruchstellern war Anlass dafür, dieses System zu hinterfragen. Obwohl die Konformitätsbewertung entsprechend der skizzierten Regeln erteilt worden war, sind nach wie vor weltweit unzählige Prozesse von Geschädigten anhängig, die sich – wohl auch in Ermangelung einer ausreichenden Haftpflichtdeckung des mit krimineller Energie handelnden Herstellers – gegen die Benannte Stelle richten.14
Grundsätzlich richtet sich auch die Produkthaftung für Medizinprodukte nach den zu § 823 BGB für die Produzentenhaftung entwickelten Grundsätzen und dem Produkthaftungsgesetz.15
Während die deliktische Produzentenhaftung grundsätzlich ein Verschulden des Anspruchsgegners voraussetzt, ist die Produkthaftung eine verschuldensunabhängige Gefährdungshaftung. Die Besonderheit bei der von der Rechtsprechung entwickelten Produzentenhaftung ist die Beweislastumkehr: Da der Geschädigte selbst wenig Einsicht in die Vorgänge hat, die sich bei der Herstellung des Produkts im Betrieb abgespielt haben, und er daher nur schwer seinen Anspruch darlegen und beweisen kann, muss der Schädiger beweisen, dass ihn kein Verschulden trifft, er also keine Verkehrspflicht verletzt hat. Eine Verkehrspflicht ergibt sich für den Hersteller daraus, dass derjenige, der ein Produkt herstellt und in den Verkehr bringt, eine Gefahrenquelle schafft. Zudem muss der Hersteller beweisen, dass keines seiner Organe nach § 31 BGB eine Verkehrspflicht verletzt hat. Der Geschädigte hingegen muss beweisen, dass ein Produktfehler vorliegt.16 Dabei ist der Fehlerbegriff des ProdHaftG mit dem Fehlerbegriff der Produzentenhaftung identisch.17
Bei der Konzeption des ProdHaftG ist der Einfluss europäischer Rechtsnormen nicht zu unterschätzen. Das Gesetz setzt die Vorgaben der Produkthaftungsrichtlinie18 um. Zusätzlich wird mit der unten noch näher dargestellten Medizinprodukteverordnung19 eine eigenständige Anspruchsgrundlage geschaffen. So heißt es in Art. 10 Abs. 16 MDR:
„Natürliche oder juristische Personen können für einen Schaden, der durch ein fehlerhaftes Produkt verursacht wurde, gemäß dem geltenden Unionsrecht und dem geltenden nationalen Recht Schadensersatz verlangen.“
Eine solche Vorschrift gab es in der Medizinprodukterichtlinie nicht.20
Die europarechtlichen Regelungen erweitern auch den Kreis der Haftungssubjekte in diesem Rechtsgebiet. Neben den „üblichen“ möglichen Schuldnern nach dem ProdHaftG, nämlich dem Hersteller/Zulieferer und dem Importeur, kam bisher lediglich noch die Benannte Stelle unter den beschriebenen engen Voraussetzungen als Haftende in Betracht.21 Zukünftig wird auch der sog. „Bevollmächtigte“ des Herstellers in Anspruch genommen werden können.22
Aufgrund des großen Einflusses europäischer Verordnungen und Richtlinien ist der Vorlagebeschluss des BGH im Fall der mit Industriesilikon gefüllten Implantate nicht der einzige. Exemplarisch sei in diesem Zusammenhang der recht bekannt gewordene Fall skizziert, bei dem eine Krankenkasse vom Hersteller von Herzschrittmachern die Operationskosten der Erst-OP und die Austauschkosten (Zweit-OP) forderte, nachdem dieser mitgeteilt hatte, dass es zu einem Ausfall der Geräte ohne Vorwarnung kommen könne.23
Auch hier setzte der BGH zunächst das Verfahren aus und legte dem EuGH die Sache vor, um den Fehlerbegriff aus Art. 6 der Produkthaftungsrichtlinie24 für den Bereich der Medizinprodukte klären zu lassen.25 Nach dieser Vorschrift ist ein Produkt fehlerhaft, „wenn es nicht die Sicherheit bietet, die man unter Berücksichtigung aller Umstände ... zu erwarten berechtigt ist.“
Im konkreten Fall stellte sich die Frage, ob i. d. Sinne ein in den menschlichen Körper implantiertes Medizinprodukt bereits dann fehlerhaft ist, wenn Produkte derselben Produktgruppe ein nennenswert erhöhtes Ausfallrisiko haben, ein Fehler bei dem im konkreten Fall implantierten Gerät aber nicht festgestellt ist. Handelt es sich bei den Kosten der Operation zur Explantation des Produkts und zur Implantation eines anderen Herzschrittmachers um einen durch Körperverletzung verursachten Schaden i. S. der Richtlinie?
Der EuGH beantwortete die Fragen dahingehend, dass bei medizinischen Geräten die Anforderungen an ihre Sicherheit besonders hoch anzusetzen sind und daher die Richtlinie dahin auszulegen sei, dass ein Medizinprodukt, das zu einer Gruppe oder Serie gehöre, bei denen ein potenzieller Fehler festgestellt wurde, als fehlerhaft eingestuft werden kann, ohne dass der Fehler bei dem konkreten Produkt festgestellt werden muss.26 Auch der Schadensbegriff aus Art. 9 Satz 1a der Richtlinie sei i. d. Sinne weit auszulegen und umfasse „alles, was erforderlich ist, um die Schadensfolgen zu beseitigen … auch die Kosten im Zusammenhang mit dem Austausch des fehlerhaften Produkts“.
Im Rahmen der Medizinprodukthaftung ist daher festzuhalten: Bereits das Risiko eines Mangels kann Auslöser für die Produkthaftung sein. Die tatsächliche Fehlerhaftigkeit des Produkts ist nicht relevant.
Eine mögliche Grenze der Erstattungspflicht könnte dabei die Entscheidung des BGH vom 16. Dezember 2008 darstellen.27 In diesem Fall hatte eine Krankenkasse elektrisch verstellbare Pflegebetten gekauft, bei denen aufgrund des möglichen Eindringens von Feuchtigkeit in den Verstellmechanismus eine Brandgefahr bestand. Während der Hersteller einen Nachrüstsatz einschließlich Einbau für DEM 350 bis DEM 400 je Bett anbot, verlangte die Krankenkasse eine Nachrüstung ohne Berechnung.
Der BGH lehnte eine Pflicht des Herstellers zur kostenlosen Nachrüstung ab. Diese bestehe nur unter besonderen Voraussetzungen, nämlich einer konkreten Gefahr für die durch § 823 Abs. 1 BGB geschützten Rechtsgüter.28 Sind hingegen bereits andere – weniger weitreichende – Maßnahmen zur Gefahrenabwehr geeignet,29 ist der Hersteller nicht verpflichtet, ein fehlerfreies Produkt zur Verfügung zu stellen.30
Vereinfacht: Solange ohne den Austausch keine Lebensgefahr besteht und schon die bloße Nichtnutzung die Gefahr beseitigt, besteht auch keine aus dem ProdHaftG resultierende Nachrüstungs- und Reparaturpflicht des Herstellers.
Wie schon mehrfach dargestellt, sollte die MDR31 die als unzureichend empfundene Haftungssituation verbessern und sowohl das Zulassungsverfahren als auch die Haftung grundlegend reformieren und damit verbessern. Wie ist dieses Vorhaben nun konkret umgesetzt worden?
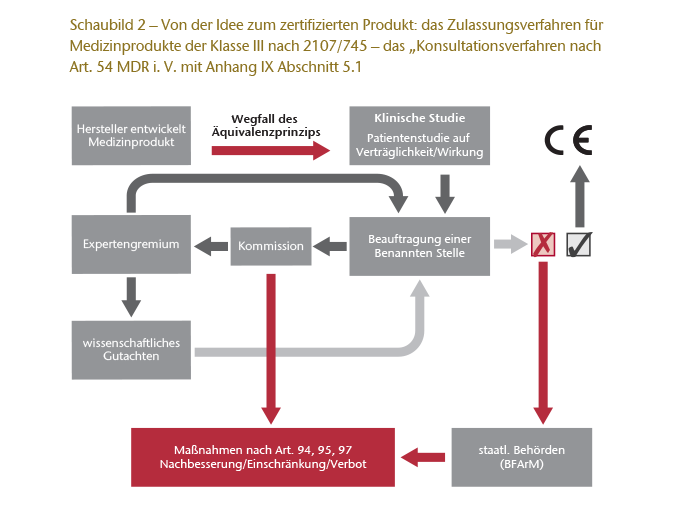
Mit Inkrafttreten der MDR wird ein neues „Konsultationsverfahren“ (engl. „scrutiny“ oder „consultation procedure“ (Art. 54 ff. MDR)) eingeführt. Zwar werden nach wie vor die Benannten Stellen Partner/Begleiter im Zertifizierungsprozess sein, jedoch muss die Benannte Stelle nun der Kommission die Bewertung einer vom Hersteller durchgeführten klinischen Studie weiterleiten. Die Kommission „konsultiert“ sodann ein Expertengremium, das die Studie und die Bewertung der Benannten Stelle im Rahmen eines wissenschaftlichen Gutachtens überprüft. Fällt das Gutachten positiv aus, kann die „Benannte Stelle“ die Konformitätserklärung abgeben. Anderenfalls muss sie Nachbesserungen veranlassen. Eine negative Bewertung kann aber auch anstatt einer Nachbesserung zu einer Einschränkung oder gar einem Verbot der Zulassung führen.32
Darüber hinaus sind in der neuen Verordnung Neuregelungen zur Marktüberwachung mit kürzeren Meldefristen enthalten und die Anforderungen an das Qualitätsmanagement-System sowie die technische Dokumentation wurden erhöht.
Am wichtigsten dürfte jedoch sein, dass mit diesem Konsultationsverfahren die bisher so umstrittene Zulassung nach dem Äquivalenzprinzip faktisch wegfällt. Vielmehr ist nahezu immer eine eigene klinische Prüfung vorzulegen. Die Äquivalenzbetrachtung ist nur noch dann möglich, wenn man zum Nachweis der Gleichartigkeit des neu zuzulassenden Produkts auf vergleichende Rohdaten eines zustimmenden Mitbewerbers zurückgreifen kann. Für Implantate und Klasse-III-Produkte werden klinische Prüfungen damit praktisch Pflicht.
Schaubild 2 zeigt das deutlich komplexere Zulassungsverfahren für Produkte der Klasse III nach Art. 54 ff. MDR i. V. mit Anhang IX Abschnitt 5.1 der MDR.
Auch die Haftungsszenarien sind durch die MDR konkretisiert und ausgeweitet worden.
Die Pflichten der Benannten Stellen im Zertifizierungsprozess finden sich im Wesentlichen in Anhang IX (dort Abschnitt 3.4.) der MDR. Neu sind in diesem Zusammenhang insbesondere die Pflicht zur verdachtsunabhängigen Durchführung unangekündigter Audits der Hersteller mindestens einmal alle fünf Jahre und die Stichprobenprüfung der zertifizierten Produkte. Gerade wegen der bereits angesprochenen33 eigenständigen Anspruchsgrundlage in Art. 10 Abs. 16 MDR muss zukünftig für das Bejahen einer Haftung der Benannten Stelle immer geprüft werden:
Auch wenn der Hersteller nach wie vor für sein Produkt verantwortlich bleibt und die Benannten Stellen weiterhin nur Begleiter in Konformitätsbewertungsverfahren sein werden, bedeutet diese Erweiterung der Pflichten, dass für die Benannten Stellen eine vertragliche Regelung der internen Haftungsverteilung zumindest für einen etwaigen Gesamtschuldnerausgleich ratsam ist.
Art. 11 der MDR regelt die Rechtsstellung des Bevollmächtigten34 und dessen Haftung35. Dies ist insbesondere für den Fall von Interesse, dass der Hersteller keine Niederlassung in der EU hat. Ohne einen Bevollmächtigen zu benennen, kann dieser nämlich nach Art. 11 Abs. 1 MDR kein Medizinprodukt in Europa in den Verkehr bringen.36 Verstößt ein außereuropäischer Hersteller gegen seine Pflichten aus Art. 10 MDR insbesondere durch eine fehlerhafte Konzeption oder Herstellung des Produkts und bringt damit ein fehlerhaftes Produkt in den Verkehr, haftet der Bevollmächtigte gem. Art. 11 Abs. 5 MDR „… auf der gleichen Grundlage wie der Hersteller … als Gesamtschuldner“. Obwohl der Bevollmächtigte typischerweise nur administrative Aufgaben wahrnimmt, trifft ihn die volle Haftung für Produktfehler trotz beschränkter Rechtsstellung. Hinzu kommt, dass bei Pflichtverletzungen des Herstellers selbst eine Beendigung des Mandats nicht zur Enthaftung des Bevollmächtigten führt (Art. 11 Abs. 3 (h) MDR).
Im Vorfeld des Erlasses der Verordnung wurde viel über die Einführung einer Versicherungspflicht für die genannten Haftungssubjekte diskutiert. Davon hat man jedoch zumindest für die Hersteller und Bevollmächtigten abgesehen. Dabei zeigt gerade der sog. PIP-Skandal deutlich, welchen Umfang etwaige Schadensersatzansprüche erreichen können.
Während Art. 10 Abs. 16, 2. Unterabschnitt der MDR die Hersteller zumindest zu einer Deckungsvorsorge verpflichtet, der sie durch entsprechend ausreichende Rückstellungen (oder optional den Nachweis einer Haftpflichtversicherung) nachkommen können, findet sich in der Verordnung keine vergleichbare Vorschrift für den Bevollmächtigen.37 Der Hersteller ist gem. Anhang XV, Kapitel 2, Abschnitt 4.3 MDR lediglich verpflichtet, für die klinische Prüfung zur Zertifizierung eine Haftpflichtversicherung für Schäden der Prüfungsteilnehmer nachzuweisen.
Jedoch sind die Benannten Stellen nach Anhang VII, Abschnitt 1.4. der MDR zum Abschluss einer Haftpflichtversicherung verpflichtet.
Das Zulassungsverfahren nach der bisherigen MDD (93/42/EWG) wurde der wachsenden Bedeutung von Medizinprodukten sicher nicht gerecht. Folgerichtig ist die Neuregelung der MDR (2107/745) deutlich komplexer (Stichwort: Scrutiny-Verfahren). Nicht zuletzt durch die erweiterten Pflichten der Benannten Stellen und die Einführung einer Haftung der Bevollmächtigten sind auch die Haftungsmöglichkeiten konkretisiert und erweitert worden, aber natürlich wird auch das Zulassungsverfahren verlängert und verteuert. Auf der anderen Seite dürfte unzweifelhaft der Verbraucherschutz gestärkt worden sein.
Insgesamt kann festgehalten werden, dass sowohl die neue MDR als auch das deutsche Haftungsrecht für Medizinprodukte von der Besonderheit geprägt sind, dass gerade bei diesen Produkten die Sicherheitsanforderungen besonders hoch anzusetzen sind. Dies wird sowohl durch den weit gefassten Fehlerbegriff deutlich als auch durch das von der deutschen Rechtsprechung im Zusammenhang mit der Haftung entwickelte Abgrenzungskriterium einer „Gefahr für Leib und Leben“.
Ob die Neuregelungen geeignet und ausreichend sind, einen solchen Schutz der Patienten zu gewährleisten, wird die Zukunft zeigen. Dennoch bleiben bereits jetzt Fragen offen. Exemplarisch seien nur zwei genannt:
Dies sind nur einige wenige Herausforderungen, denen die neue MDR bereits jetzt begegnet, die aber nicht geregelt sind.
Dennoch bleibt es für die Zertifizierung aller bestehenden Medizinprodukte nach den neuen MDR-Vorschriften bei der Frist von Mai 2020. Doch wird dies überhaupt möglich sein?
Die Verschärfung der Gesetze hat die Europäische Kommission veranlasst, alle Prüfstellen (d. h. die benannten Stellen) zu prüfen, die derzeit die CE-Zertifizierung vergeben. Da bisher als Prüfstellen nur der TÜV Süd in München und das BSI-Institut in Großbritannien zugelassen sind, können – wie es die Bundesregierung formuliert hat – „Versorgungsengpässe – Stand heute – nicht ausgeschlossen werden“.40



















